
OZEMPIC 1 mg, solution injectable SC, boîte de 1 stylo prérempli multidose ( 4 aig) de 3 ml
Dernière révision : 13/02/2025
Taux de TVA : 2.1%
Prix de vente : 76,58 €
Taux remboursement SS : 30%
Base remboursement SS : 76,58 €
Laboratoire exploitant : NOVO NORDISK
Source :

Ozempic est indiqué chez les adultes pour le traitement du diabète de type 2 insuffisamment contrôlé en complément d'un régime alimentaire et d'une activité physique
- en monothérapie, quand l'utilisation de la metformine est considérée comme inappropriée en raison d'une intolérance ou de contre-indications
- en association avec d'autres médicaments destinés au traitement du diabète.
Pour les résultats des essais concernant les associations, les effets sur le contrôle glycémique, les événements cardiovasculaires et les événements rénaux, ainsi que sur les populations étudiées, voir les rubriques Mises en garde spéciales et précautions d'emploi, Interactions avec d'autres médicaments et autres formes d'interactions et Propriétés pharmacodynamiques.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Généralités
Le sémaglutide ne doit pas être utilisé chez les patients présentant un diabète de type 1 ou pour le traitement d'une acidocétose diabétique. Le sémaglutide n'est pas un substitut de l'insuline. Des cas d'acidocétose diabétique ont été rapportés chez les patients insulino-dépendants ayant eu un arrêt rapide de leur insuline ou ayant eu une réduction de la dose de leur insuline au moment de l'initiation par un agoniste des récepteurs du GLP-1 (voir rubrique Posologie et mode d'administration).
Il n'y a pas d'expérience chez les patients présentant une insuffisance cardiaque congestive de classe IV New York Heart Association (NYHA), le sémaglutide n'est donc pas recommandé chez ces patients.
Aspiration pulmonaire en association avec une anesthésie générale ou une sédation profonde
Des cas d'aspiration pulmonaire du contenu gastrique ont été signalés chez des patients recevant des agonistes des récepteurs du GLP-1 subissant une anesthésie générale ou une sédation profonde. Par conséquent, le risque accru de contenu gastrique résiduel en raison du retard de vidange gastrique (voir section Effets indésirables) doit être pris en considération avant de réaliser des procédures impliquant une anesthésie générale ou une sédation profonde.
Effets gastro-intestinaux
L'utilisation d'agonistes des récepteurs du GLP-1 peut être associée à des réactions indésirables gastro-intestinales. Cela doit être pris en compte lors du traitement des patients présentant une altération de la fonction rénale, car les nausées, les vomissements et la diarrhée peuvent entraîner une déshydratation susceptible de détériorer la fonction rénale (voir rubrique Effets indésirables).
Pancréatite aiguë
Des cas de pancréatite aiguë ont été observés lors de l'utilisation d'agonistes des récepteurs du GLP-1. Les patients doivent être informés des symptômes caractéristiques de la pancréatite aiguë. En cas de suspicion de pancréatite, le sémaglutide devra être arrêté ; si la pancréatite est confirmée, le sémaglutide ne devra pas être réadministré. Il convient d'être prudent chez les patients ayant des antécédents de pancréatite.
Hypoglycémie
Les patients traités par le sémaglutide en association à un sulfamide hypoglycémiant ou à une insuline peuvent présenter une augmentation du risque d'hypoglycémie. Le risque d'hypoglycémie peut être diminué en réduisant la dose du sulfamide hypoglycémiant ou de l'insuline lors de l'initiation du traitement par sémaglutide (voir rubrique Effets indésirables).
Rétinopathie diabétique
Un
risque accru de complications liées à la rétinopathie diabétique a été
observé chez les patients souffrant de rétinopathie diabétique et
traités par insuline et sémaglutide (voir rubrique Effets indésirables).
Il convient d'être prudent lors de l'utilisation du sémaglutide chez
des patients souffrant de rétinopathie diabétique et traités par
insuline. Ces patients doivent faire l'objet d'un suivi attentif et
être traités selon les recommandations cliniques. Une amélioration
rapide du contrôle glycémique a été associée à une aggravation
provisoire de la rétinopathie diabétique, cependant d'autres mécanismes
ne peuvent pas être exclus.
Aucune expérience n'a été acquise avec le sémaglutide de 2 mg chez les
patients atteints de diabète de type 2 présentant une rétinopathie
diabétique non contrôlée ou potentiellement instable ; le sémaglutide
de 2 mg n'est par conséquent pas recommandé chez ces patients.
Teneur en sodium
Ce médicament contient moins de 1 mmol de sodium (23 mg) par dose, c'est-à-dire qu'il est essentiellement « sans sodium ».
Résumé du
profil de sécurité
Lors de 8 essais de phase 3a, 4 792 patients ont été exposés à 1 mg au maximum de sémaglutide. Les réactions indésirables les plus fréquemment rapportées pendant les essais cliniques étaient les affections gastro-intestinales, incluant les nausées (très fréquentes), les diarrhées (très fréquentes) et les vomissements (fréquents). En général, ces réactions étaient d'intensité légère ou modérée et de courte durée.
Liste
tabulée des réactions indésirables
Le tableau 1 répertorie les réactions indésirables rapportées lors de tous les essais de phase 3 (notamment l'étude de résultats cardiovasculaires à long terme) et dans les rapports post-commercialisation chez les patients diabétiques de type 2 (décrit plus en détail à la rubrique Propriétés pharmacodynamiques). La fréquence des réactions indésirables (excepté les complications de rétinopathie diabétique, voir note du tableau 1) repose sur un ensemble d'essais de phase 3a, excluant l'essai d'évaluation des résultats cardiovasculaires (voir le texte sous le tableau pour plus d'informations).
Les réactions sont indiquées ci-dessous par classe de systèmes d'organes et par fréquence absolue. Les fréquences sont définies comme suit : très fréquent : (≥ 1/10) ; fréquent : (≥ 1/100, < 1/10) ; peu fréquent : (≥ 1/1 000, < 1/100) ; rare : (≥ 1/10 000, < 1/1 000) ; très rare : (< 1/10 000) et fréquence indéterminée : (ne peut être estimée sur la base des données disponibles). Au sein de chaque groupe de fréquence, les réactions indésirables sont présentées suivant un ordre décroissant de gravité.
Tableau 1 Fréquence des réactions indésirables du sémaglutide
|
Classe
de systèmes d'organes MedDRA |
Très fréquent | Fréquent | Peu fréquent | Rare |
Fréquence
indéterminée |
| Affections du système immunitaire | Hypersensibilitéc | Réaction anaphylactique |
|
||
| Troubles du métabolisme et de la nutrition | Hypoglycémiea en cas d'utilisation avec de l'insuline ou un sulfamide hypoglycémiant |
Hypoglycémiea en cas d'utilisation
avec d'autres antidiabétiques oraux (ADO) Diminution de l'appétit |
|
||
| Affections du système nerveux | Vertiges | Dysgueusie |
|
||
| Affections oculaires | Complications de la rétinopathie diabétiqueb |
|
|||
| Affections cardiaques | Augmentation de la fréquence cardiaque |
|
|||
| Affections gastro-intestinales |
Nausées Diarrhées |
Vomissements Douleur abdominale Distension abdominale Constipation Dyspepsie Gastrite Reflux gastro-œsophagien Éructation Flatulences |
Pancréatite
aiguë
Retard de la vidange gastrique |
Obstruction intestinaled |
|
| Affections hépatobiliaires | Lithiase biliaire | ||||
|
Affections
de la
peau et du tissu sous-cutané |
|
|
|
|
Angiœdèmed |
| Troubles généraux et anomalies au site d'administration | Fatigue | Réactions au site d'injection |
|
||
| Investigations |
Lipase
augmentée Amylase augmentée Perte de poids |
|
a) Hypoglycémie définie
comme sévère (nécessitant l'aide d'une autre personne) ou symptomatique
combinée à une glycémie < 3,1 mmol/l
b) Les complications de la rétinopathie diabétique englobent : photocoagulation rétinienne, traitement par des agents intravitreux, hémorragie vitreuse, cécité diabétique (peu fréquent). Fréquence basée sur l'essai d'évaluation des résultats cardiovasculaires.
c) Groupement de terme couvrant également les événements indésirables liés à l'hypersensibilité tels que les éruptions cutanées et l'urticaire.
d) D'après les rapports post-commercialisation.
b) Les complications de la rétinopathie diabétique englobent : photocoagulation rétinienne, traitement par des agents intravitreux, hémorragie vitreuse, cécité diabétique (peu fréquent). Fréquence basée sur l'essai d'évaluation des résultats cardiovasculaires.
c) Groupement de terme couvrant également les événements indésirables liés à l'hypersensibilité tels que les éruptions cutanées et l'urticaire.
d) D'après les rapports post-commercialisation.
Essai de
sécurité et d'évaluation des résultats cardiovasculaires sur 2 ans
Dans une population à haut risque cardiovasculaire, le profil des réactions
indésirables était similaire à celui observé dans les autres essais de phase 3a
(décrits à la rubrique Propriétés pharmacodynamiques).
Description de certaines réactions indésirables
Hypoglycémie
Aucun épisode d'hypoglycémie sévère n'a été observé lorsque le sémaglutide était utilisé en monothérapie. Les
hypoglycémies sévères ont principalement été observées lorsque le sémaglutide était associé à un sulfamide hypoglycémiant
(1,2 % des patients, 0,03 événement/patient-année) ou à de l'insuline (1,5 %
des patients, 0,02 événement/patient-année). Peu d'épisodes d'hypoglycémie (0,1
% des patients, 0,001 événement/patient-année) ont été observés lors de
l'administration du sémaglutide en association à des
antidiabétiques oraux autres que les sulfamides hypoglycémiants.
Une hypoglycémie selon l'American Diabetes Association (ADA, assosciation américaine du diabète) est survenue chez 11,3 % (0,3 événement/patient-année) des patients lorsque le sémaglutide 1 mg était ajouté à un inhibiteur du SGLT2 dans SUSTAIN 9 versus 2,0 % (0,04 événement/patient-année) des patients recevant le placebo. Une hypoglycémie sévère a été rapportée chez respectivement 0,7 % (0,01 événement/patient-année) et 0 % des patients.
Dans un essai de phase 3b de 40 semaines mené chez des patients
recevant 1 mg et 2 mg de sémaglutide, la majorité des épisodes
hypoglycémiques (45 épisodes sur 49) est survenue lorsque le
sémaglutide était utilisé en association avec un sulfamide
hypoglycémiant ou de l'insuline. Globalement, il n'existe pas de risque
accru d'hypoglycémie avec le sémaglutide de 2 mg.
Réactions
indésirables gastro-intestinales
Des nausées sont survenues chez 17 % et 19,9 % des patients lorsqu'ils
étaient traités avec respectivement 0,5 mg et 1 mg de sémaglutide ;
des diarrhées sont apparues chez respectivement 12,2 % et 13,3 % des patients
et des vomissements chez respectivement 6,4 % et 8,4 % des patients.
La plupart de ces événements étaient d'intensité légère à modérée et de courte
durée. Les événements ont entraîné un arrêt du traitement chez respectivement
3,9 % et 5 % des patients. Les événements étaient plus fréquemment rapportés
pendant les premiers mois de traitement.
Les patients de faible poids corporel peuvent être davantage sujets aux effets
indésirables gastro-intestinaux lorsqu'ils sont traités par sémaglutide.
Dans un essai de phase 3b de 40 semaines mené chez des patients recevant 1 mg et 2 mg de sémaglutide, les nausées sont survenues en proportion similaire chez les patients traités par le sémaglutide de 1 mg et de 2 mg, respectivement. Les diarrhées et les vomissements sont survenues chez une proportion plus élevée de patients traités par le sémaglutide de 2 mg que de patients traités par le sémaglutide de 1 mg. Les réactions indésirables gastro-intestinales ont entrainé un arrêt du traitement en proportion similaire dans les groupes de traitement par le sémaglutide de 1 mg et de 2 mg.
En
utilisation concomitante avec un inhibiteur du SGLT2 dans SUSTAIN 9,
une constipation et un reflux gastro-œsophagien sont survenus
respectivement chez 6,7 % et 4 % des patients traités par sémaglutide
1mg versus aucun événement chez les patients recevant le placebo. La
prévalence de ces événements n'a pas diminué avec le temps.
Pancréatites
aiguës
La fréquence rapportée des pancréatites aiguës confirmées par adjudication dans
les études cliniques de phase 3a était respectivement de 0,3 % pour le sémaglutide et 0,2 % pour le comparateur. Dans l'essai
d'évaluation des résultats cardiovasculaires sur 2 ans, la fréquence des
pancréatites aiguës confirmées par adjudication était de 0,5 % pour le sémaglutide et de 0,6 % pour le placebo (voir rubrique
Mises en garde spéciales et précautions d'emploi).
Complications
liées à la rétinopathie diabétique
Un essai clinique sur 2 ans a étudié 3 297 patients diabétiques de type 2, avec
un risque cardiovasculaire élevé, un diabète ancien et un contrôle glycémique
insatisfaisant. Lors de cet essai, des événements de complications liées à la
rétinopathie diabétique confirmés par adjudication sont survenus chez plus de
patients traités par sémaglutide (3 %) comparé à
ceux sous placebo (1,8 %). Cela a été observé chez des patients insulino-traités avec une rétinopathie diabétique connue.
La différence de traitement est apparue rapidement et a persisté tout au long
de l'essai. L'évaluation systématique des complications liées à la rétinopathie
diabétique n'a été réalisée que dans l'essai d'évaluation des résultats
cardiovasculaires. Lors d'essais cliniques d'une durée allant jusqu'à un an et
portant sur 4 807 patients diabétiques de type 2, les événements indésirables
liés à la rétinopathie diabétique ont été rapportés dans des proportions
similaires chez des patients traités par sémaglutide
(1,7 %) et par les comparateurs (2,0 %).
Arrêt dû
à un événement indésirable
L'incidence de l'arrêt du traitement dû à des événements indésirables était de
6,1 % et 8,7 % chez les patients recevant respectivement 0,5 mg et 1 mg de sémaglutide, versus 1,5 % dans le groupe placebo. Les
événements indésirables entraînant le plus fréquemment un arrêt du traitement
étaient de nature gastro-intestinale.
Réactions
au site d'injection
Des réactions au site d'injection (par exemple rash au site d'injection,
érythème) ont été rapportées par 0,6 % et 0,5 % des patients sous 0,5 mg et 1
mg de sémaglutide respectivement. Ces réactions
étaient généralement de faible intensité.
Immunogénicité
Compte tenu des propriétés potentiellement immunogènes des médicaments
contenant des protéines ou des peptides, les patients traités par sémaglutide peuvent développer des anticorps. La proportion
de patients testés positifs aux anticorps anti-sémaglutide
à tout moment après l'inclusion était faible (1-3 %) et aucun patient ne
présentait d'anticorps neutralisants anti-sémaglutide
ni d'anticorps anti-sémaglutide avec un effet
neutralisant du GLP-1 endogène à la fin de l'essai.
Augmentation de la fréquence cardiaque
Une augmentation de la fréquence cardiaque a été observée avec les
agonistes
des récepteurs du GLP-1. Dans les essais de phase 3a, des augmentations
moyennes
de 1 à 6 battements par minute (bpm) par rapport à
une valeur initiale de 72 à 76 bpm ont été observées
chez les patients traités par Ozempic. Dans un essai
à long terme sur des patients présentant des facteurs de risque
cardiovasculaires, 16 % des patients traités par Ozempic
ont présenté une augmentation de la fréquence cardiaque supérieure à 10
bpm, contre 11 % des patients sous placebo après 2 ans de
traitement.
Déclaration
des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via l'Agence nationale de sécurité du médicament et des produits de santé (ANSM) et le réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
DEMANDER UNE AIDE MEDICALE ET INFORMER LE MEDECIN en cas de réaction allergique : problèmes respiratoires, gonflement de la gorge et du visage, augmentation du rythme cardiaque.
CONSULTER IMMEDIATEMENT le médecin en cas de douleurs intenses et persistantes au niveau de l'estomac..
INFORMER le médecin en cas de problèmes au niveau des yeux, tels que des modifications de la vue.
CONTRACEPTION :
- L'utilisation d'une CONTRACEPTION est recommandée chez les femmes en âge de procréer.
- Le traitement par sémaglutide DOIT ÊTRE ARRETE au moins 2 mois avant un projet de grossesse.
PRUDENCE en cas de conduite de véhicule ou d'utilisation de machines (risque d'hypoglycémie).
INFORMER le médecin en cas d'intervention chirurgicale nécessitant une anesthésie (endormissement).
Femmes en âge de procréer
L'utilisation d'une contraception pendant le traitement par sémaglutide est recommandée chez les femmes en âge de procréer.
Grossesse
Les études effectuées chez l'animal ont mis en évidence une toxicité sur la reproduction (voir rubrique Données de sécurité préclinique). Il existe des données limitées sur l'utilisation du sémaglutide chez la femme enceinte. Le sémaglutide ne doit donc pas être utilisé pendant la grossesse. En cas de projet de grossesse ou en cas de grossesse, le traitement par sémaglutide doit être interrompu. Le sémaglutide doit être arrêté au moins 2 mois avant un projet de grossesse en raison de sa longue demi-vie (voir rubrique Propriétés pharmacocinétiques).
Allaitement
Le sémaglutide a été excrété dans le lait de rates allaitantes. Un risque pour l'enfant allaité ne pouvant être exclu, le sémaglutide ne doit pas être utilisé pendant l'allaitement.
Fertilité
L'effet du sémaglutide sur la fertilité humaine est inconnu. Le sémaglutide n'a pas affecté la fertilité des rats mâles. Chez le rat femelle, une prolongation du cycle œstrien et une faible baisse du nombre d'ovulations ont été observées à des doses associées à une réduction du poids maternel (voir rubrique Données de sécurité préclinique).
Le sémaglutide retarde la vidange gastrique et est susceptible d'influencer le taux d'absorption des médicaments administrés par voie orale de façon concomitante. Le sémaglutide doit être utilisé avec prudence chez les patients recevant des médicaments par voie orale nécessitant une absorption gastro-intestinale rapide.
Paracétamol
Le sémaglutide retarde la vidange gastrique telle qu'évaluée par la pharmacocinétique du paracétamol pendant un repas test standard. L'ASC0-60 min et la Cmax du paracétamol ont baissé de 27 % et 23 % respectivement, après une utilisation concomitante de 1 mg de sémaglutide. L'exposition totale au paracétamol (ASC0-5 h) n'a pas été affectée. Aucun effet cliniquement pertinent sur la vitesse de vidange gastrique n'a été observé avec le sémaglutide de 2,4 mg, après 20 semaines d'administration du sémaglutide, probablement en raison d'un effet de tolérance. Aucun ajustement de la dose de paracétamol n'est nécessaire en cas d'association au sémaglutide.
Contraceptifs oraux
Le sémaglutide ne devrait pas réduire l'effet des contraceptifs oraux, car le sémaglutide ne modifie pas de manière cliniquement significative l'exposition totale à l'éthinylestradiol et au lévonorgestrel en cas d'administration concomitante d'un contraceptif oral combiné (0,03 mg d'éthinylestradiol/0,15 mg de lévonorgestrel) avec le sémaglutide. L'exposition à l'éthinylestradiol n'a pas été affectée ; une augmentation de 20 % de l'exposition au lévonorgestrel à l'état d'équilibre a été observée. La Cmax n'a été affectée pour aucun des composés.
Atorvastatine
Le sémaglutide n'a pas modifié l'exposition totale à l'atorvastatine après administration d'une dose unique de 40 mg d'atorvastatine. La Cmax de l'atorvastatine a diminué de 38 %. Cette baisse n'a pas été considérée comme cliniquement significative.
Digoxine
Le sémaglutide n'a pas modifié l'exposition totale ou la Cmax de la digoxine après administration d'une dose unique de 0,5 mg de digoxine.
Metformine
Le sémaglutide n'a pas modifié l'exposition totale ou la Cmax de la metformine après administration de 500 mg deux fois par jour pendant 3,5 jours.
Warfarine et autres dérivés de la coumarine
Le sémaglutide n’a pas modifié l’exposition totale ou la Cmax
de la R- et S-warfarine après une dose unique de warfarine (25 mg), et
l’effet pharmacodynamique de la warfarine tel que mesuré par le rapport
normalisé international (INR) n’a pas été affecté de manière
cliniquement significative.
Toutefois, des cas de diminution de l'INR ont été rapportés lors de
l'utilisation concomitante d'acénocoumarol et de sémaglutide. Lors de
l’initiation du traitement par sémaglutide chez des patients sous
warfarine ou autres dérivés de la coumarine, il est recommandé de
surveiller fréquemment l’INR.
Posologie
La dose initiale est de 0,25 mg de sémaglutide une fois par semaine. Après 4 semaines de traitement, la dose devra être augmentée à 0,5 mg une fois par semaine. Après au moins 4 semaines à une dose de 0,5 mg une fois par semaine, la dose peut être augmentée à 1 mg une fois par semaine pour améliorer davantage le contrôle glycémique. Après au moins 4 semaines à une dose de 1 mg une fois par semaine, la dose peut être augmentée à 2 mg une fois par semaine pour améliorer davantage le contrôle glycémique.
La dose de 0,25 mg de sémaglutide n'est pas une dose d'entretien. Des doses hebdomadaires supérieures à 2 mg ne sont pas recommandées.
Lorsqu'Ozempic est ajouté à un traitement existant par metformine et/ou thiazolidinedione ou à un inhibiteur du co-transporteur de sodium-glucose de type 2 (SGLT2), le traitement par metformine et/ou thiazolidinedione ou inhibiteur du SGLT2 peut être poursuivi à la même dose.
Lorsqu'Ozempic est ajouté à un traitement existant par sulfamide hypoglycémiant ou par insuline, une diminution de la dose du sulfamide hypoglycémiant ou de l'insuline devra être envisagée afin de réduire le risque d'hypoglycémie (voir rubriques Mises en garde spéciales et précautions d'emploi et Effets indésirables).
Une autosurveillance glycémique n'est pas nécessaire pour ajuster la dose d'Ozempic. Une autosurveillance glycémique est nécessaire afin d'ajuster la dose du sulfamide hypoglycémiant ou d'insuline, particulièrement au moment de l'initiation de Ozempic et de la réduction de la dose d'insuline. Une diminution progressive de l'insuline est recommandée.
Oubli de dose
Si une dose est oubliée, elle doit être administrée dès que possible et
dans les 5 jours suivant l'oubli. Si plus de 5 jours se sont écoulés,
la dose oubliée ne doit pas être prise, et la dose suivante doit être
administrée le jour normalement prévu. Dans chacun des cas, les
patients peuvent ensuite reprendre leur schéma posologique hebdomadaire
habituel.
Modification du jour d'administration
Le jour de l'administration hebdomadaire peut être changé si
nécessaire, à condition que le délai entre deux doses soit d'au moins 3
jours (> 72 heures). Après avoir choisi un nouveau jour
d'administration, il faut continuer d'administrer la dose une fois par
semaine.
Populations particulières
Sujets âgés
Aucun ajustement de la dose n'est nécessaire en fonction de l'âge.
Insuffisance rénale
Aucun
ajustement de la dose n’est nécessaire chez les patients présentant une
insuffisance rénale légère, modérée ou sévère. L’expérience relative à
l’utilisation du sémaglutide chez les patients présentant une
insuffisance rénale au stade terminal est limitée.
Insuffisance hépatique
Aucun ajustement de la dose n'est requis chez les patients présentant
une insuffisance hépatique. L'expérience relative à l'utilisation du
sémaglutide chez des patients présentant une insuffisance hépatique
sévère est limitée. Il convient d'être prudent lors du traitement de
ces patients avec le sémaglutide (voir rubrique Propriétés pharmacocinétiques).
Population pédiatrique
La sécurité et l'efficacité du sémaglutide chez les enfants et les
adolescents âgés de moins de 18 ans n'ont pas encore été établies.
Aucune donnée n'est disponible.
Mode d'administration
Voie sous-cutanée.
Ozempic doit être injecté par voie sous-cutanée dans l'abdomen, la cuisse ou le haut du bras. Le site d'injection peut être modifié sans ajustement de la dose. Ozempic ne doit pas être administré par voie intraveineuse ou intramusculaire.
Ozempic doit être administré une fois par semaine, quel que soit le moment de la journée, au cours ou en dehors des repas.
Pour les instructions plus détaillées concernant l'administration, voir la rubrique Précautions particulières d'élimination et de manipulation.
Durée de conservation :
Avant la première utilisation
3 ans.
Après la première ouverture
Durée de conservation en cours d'utilisation : 6 semaines.
À conserver à une température ne dépassant pas 30°C ou au réfrigérateur (entre 2 °C et 8 °C). Ne pas congeler Ozempic. Conserver le capuchon sur le stylo lorsqu'il n'est pas utilisé, afin de le protéger de la lumière.
Précautions particulières de conservation :
À conserver au réfrigérateur (entre 2 °C et 8 °C). Maintenir à distance de l'élément de refroidissement. Ne pas congeler Ozempic.
Conserver le capuchon sur le stylo, afin de le protéger de la lumière.
Pour les conditions de conservation du médicament après première ouverture, voir la rubrique Durée de conservation.
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
Des cas de surdosage allant jusqu'à 4 mg en dose unique, et jusqu'à 4 mg en une semaine ont été rapportés lors des essais cliniques. La réaction indésirable la plus fréquemment rapportée était la nausée. Tous les patients se sont rétablis sans complications.
Il n'existe aucun antidote spécifique à un surdosage de sémaglutide. En cas de surdosage, un traitement symptomatique approprié doit être initié en fonction des signes cliniques et des symptômes du patient. Une période d'observation prolongée et un traitement de ces symptômes peuvent être nécessaire, en tenant compte de la longue demi-vie du sémaglutide d'environ 1 semaine (voir rubrique Propriétés pharmacocinétiques).
Classe pharmacothérapeutique : Médicaments utilisés dans le diabète, analogue du glucagon-like peptide 1 (GLP-1), Code ATC : A10BJ06
Mécanisme d'action
Le sémaglutide est un analogue du
GLP-1 présentant 94 % d’homologie avec le GLP-1 humain. Le sémaglutide
agit comme agoniste des récepteurs du GLP-1, qui se lie sélectivement
et active le récepteur du GLP-1, la cible du GLP-1 natif.
Le GLP-1 est une hormone physiologique exerçant plusieurs effets sur la régulation du glucose et de l’appétit, sur le système cardiovasculaire et les reins. Les effets sur le glucose et l’appétit sont spécifiquement médiés via les récepteurs du GLP-1 dans le pancréas et le cerveau.
Le sémaglutide réduit la glycémie de façon glucose-dépendante en stimulant la sécrétion d’insuline et en réduisant la sécrétion de glucagon lorsque la glycémie est élevée. Le mécanisme de réduction de la glycémie entraîne également un léger retard de la vidange gastrique en début de phase postprandiale. Lors d’une hypoglycémie, le sémaglutide diminue la sécrétion d’insuline sans altérer la sécrétion du glucagon.
Le sémaglutide réduit le poids corporel et la masse grasse en diminuant les apports énergétiques ; entrainant une réduction générale de l’appétit. En outre, le sémaglutide réduit la préférence pour les aliments à forte teneur en graisse.
Les récepteurs du GLP-1 sont également exprimés dans le cœur, le système vasculaire, le système immunitaire et les reins. Le mécanisme d’action du sémaglutide est probablement multifactoriel. Des effets indirects sont indiqués par l’effet bénéfique du sémaglutide sur les lipides plasmatiques, la baisse de la pression artérielle systolique et la réduction de l’inflammation selon des études cliniques, mais des effets directs sont probablement aussi impliqué. Lors d’études réalisées chez l’animal, le sémaglutide atténue le développement de l’athérosclérose en empêchant la progression de la plaque aortique et en réduisant l’inflammation dans la plaque.
Des données cliniques ont montré que le sémaglutide diminue l’albuminurie chez les patients atteints de maladie rénale.
Le GLP-1 est une hormone physiologique exerçant plusieurs effets sur la régulation du glucose et de l’appétit, sur le système cardiovasculaire et les reins. Les effets sur le glucose et l’appétit sont spécifiquement médiés via les récepteurs du GLP-1 dans le pancréas et le cerveau.
Le sémaglutide réduit la glycémie de façon glucose-dépendante en stimulant la sécrétion d’insuline et en réduisant la sécrétion de glucagon lorsque la glycémie est élevée. Le mécanisme de réduction de la glycémie entraîne également un léger retard de la vidange gastrique en début de phase postprandiale. Lors d’une hypoglycémie, le sémaglutide diminue la sécrétion d’insuline sans altérer la sécrétion du glucagon.
Le sémaglutide réduit le poids corporel et la masse grasse en diminuant les apports énergétiques ; entrainant une réduction générale de l’appétit. En outre, le sémaglutide réduit la préférence pour les aliments à forte teneur en graisse.
Les récepteurs du GLP-1 sont également exprimés dans le cœur, le système vasculaire, le système immunitaire et les reins. Le mécanisme d’action du sémaglutide est probablement multifactoriel. Des effets indirects sont indiqués par l’effet bénéfique du sémaglutide sur les lipides plasmatiques, la baisse de la pression artérielle systolique et la réduction de l’inflammation selon des études cliniques, mais des effets directs sont probablement aussi impliqué. Lors d’études réalisées chez l’animal, le sémaglutide atténue le développement de l’athérosclérose en empêchant la progression de la plaque aortique et en réduisant l’inflammation dans la plaque.
Des données cliniques ont montré que le sémaglutide diminue l’albuminurie chez les patients atteints de maladie rénale.
Effets pharmacodynamiques
Toutes les évaluations pharmacodynamiques ont été effectuées au bout de 12 semaines de traitement (incluant l'augmentation de la dose) à l'état d'équilibre avec 1 mg de sémaglutide une fois par semaine.
Glycémie
à jeun et postprandiale
Le sémaglutide réduit les concentrations de glucose à jeun et postprandiales.
Chez les patients diabétiques de type 2, le traitement par 1 mg de sémaglutide
a entraîné une réduction de la concentration de glucose en termes de variation
absolue par rapport à l'inclusion (mmol/l) et de réduction relative par rapport
au placebo (%) des concentrations de glucose à jeun (1,6 mmol/l ; réduction de
22 %), des concentrations de glucose postprandiales à 2 heures (4,1 mmol/l ;
réduction de 37 %), de la concentration de glucose moyenne sur 24 heures (1,7
mmol/l ; réduction de 22 %) et des variations de glucose postprandiales sur 3
repas (0,6-1,1 mmol/l) comparé au placebo. Le sémaglutide a réduit la glycémie
à jeun après la première dose.
Fonction
bêta-cellulaire et sécrétion d'insuline
Le sémaglutide améliore la fonction bêta-cellulaire. En comparaison avec le
placebo, le sémaglutide a amélioré la réponse insulinique de première et
deuxième phases en la multipliant par trois et par deux, respectivement, et a
augmenté la capacité de sécrétion bêta-cellulaire maximale chez les patients
diabétiques de type 2. De plus, le traitement par sémaglutide a augmenté les
concentrations d'insuline à jeun en comparaison avec le placebo.
Sécrétion
de glucagon
Le sémaglutide réduit les concentrations de glucagon à jeun et postprandiales.
Chez les patients diabétiques de type 2, le sémaglutide a entraîné les
réductions relatives suivantes du glucagon en comparaison avec le placebo :
glucagon à jeun (8-21 %), réponse postprandiale du glucagon (14- 15 %) et
concentration moyenne de glucagon sur 24 heures (12 %).
Sécrétion
de glucagon et d'insuline glucose-dépendante
Le sémaglutide a abaissé les concentrations élevées de glucose sanguin en stimulant
la sécrétion d'insuline et en réduisant la sécrétion de glucagon de façon
glucose-dépendante. Avec le sémaglutide, le taux de sécrétion d'insuline chez
les patients diabétiques de type 2 était comparable à celui des sujets sains.
Pendant l'hypoglycémie induite et en comparaison avec le placebo, le sémaglutide n'a pas modifié la réponse contre-régulatoire d'augmentation du glucagon et n'a pas limité la baisse du peptide C chez les patients diabétiques de type 2.
Vidange
gastrique
Le sémaglutide a entraîné un léger retard de la vidange gastrique au début de
la phase postprandiale, réduisant ainsi la vitesse d'apparition du glucose dans
la circulation après le repas.
Appétit,
apport énergétique et choix des aliments
En comparaison avec le placebo, le sémaglutide a diminué de 18 à 35 % l'apport
énergétique de 3 repas ad libitum consécutifs. Cette constatation a été
étayée par une suppression de l'appétit induite par le sémaglutide à jeun et en
phase postprandiale, un meilleur contrôle de l'alimentation, une diminution des
envies alimentaires et une préférence relativement réduite pour les aliments à
haute teneur en graisse.
Lipides
à jeun et postprandiaux
En comparaison avec le placebo, le sémaglutide a diminué les concentrations de
triglycérides et de cholestérol VLDL (lipoprotéines de très basse densité) à
jeun de 12 % et 21 %, respectivement. La réponse postprandiale des
triglycérides et du cholestérol VLDL à un repas à haute teneur en graisse a été
réduite de > 40 %.
Électrophysiologie
cardiaque (QTc)
L'effet du sémaglutide sur la repolarisation cardiaque a été testé lors d'un
essai QTc approfondi. Le sémaglutide n'a pas prolongé les intervalles QTc à des
niveaux de doses allant jusqu'à 1,5 mg à l'état d'équilibre.
Efficacité et sécurité cliniques
L’amélioration du contrôle glycémique, la réduction de la morbidité et de la mortalité cardiovasculaires, la perte de poids et la réduction du risque de progression de la maladie rénale chronique font partie intégrante du traitement du diabète de type 2.
L'efficacité et la sécurité de doses de 0,5 mg et de 1 mg de sémaglutide administrées une fois par semaine ont été évaluées lors de six essais randomisés contrôlés de phase 3a portant sur 7 215 patients diabétiques de type 2 (dont 4 107 traités par sémaglutide). Dans cinq essais (SUSTAIN 1-5) le critère principal était l'évaluation de l'efficacité glycémique, et dans un essai (SUSTAIN 6), le critère principal portait sur les résultats de l'évaluation cardiovasculaire.
L'efficacité et la sécurité du sémaglutide de 2 mg une fois par semaine ont été évaluées dans une étude de phase 3b (SUSTAIN FORTE) incluant 961 patients.
De plus, un essai de phase 3b (SUSTAIN 7) incluant 1 201 patients a été réalisé pour comparer l'efficacité et la sécurité du sémaglutide de 0,5 mg et de 1 mg une fois par semaine au dulaglutide 0,75 mg et 1,5 mg une fois par semaine, respectivement. Un essai de phase 3b (SUSTAIN 9) a été réalisé pour étudier l'efficacité et la sécurité du sémaglutide en complément d'un traitement par inhibiteur du SGLT2.
Le traitement par sémaglutide a démontré une réduction durable, statistiquement supérieure et cliniquement significative de l'HbA1c et du poids corporel jusqu'à 2 ans par rapport au placebo et aux traitements actifs de contrôle (sitagliptine, insuline glargine, exénatide à libération prolongée et dulaglutide).
L'efficacité du sémaglutide n'a pas été influencée par l'âge, le genre, l'origine ethnique, l'IMC à l'inclusion, le poids corporel (kg) à l'inclusion, l'ancienneté du diabète ni le niveau d'atteinte de la fonction rénale.
Les résultats ont ciblé la période sous traitement de tous les patients randomisés (les analyses étaient basées sur les modèles mixtes à mesures répétées ou à imputation multiple).
De plus, un essai de phase 3b (SUSTAIN 11) a été mené afin d'étudier l'effet du sémaglutide versus l'insuline asparte, tous deux en association à la metformine et à l'insuline glargine à dose optimale (100 U).
Un essai de phase 3b portant sur
l’évaluation des effets rénaux (FLOW), portant sur 3 533 patients, a
été mené afin d’étudier les effets du sémaglutide 1 mg une fois par
semaine versus placebo sur la progression de la maladie rénale chez les
patients atteints de diabète de type 2 et de maladie rénale chronique.
Des informations détaillées sont fournies ci-dessous.
SUSTAIN
1 - Monothérapie
Lors d'un essai clinique contrôlé de 30 semaines en double aveugle versus
placebo, 388 patients insuffisamment contrôlés par le régime alimentaire et
l'activité physique ont été randomisés dans des groupes recevant 0,5 mg ou 1 mg
de sémaglutide une fois par semaine ou un placebo.
Tableau 2 SUSTAIN 1 : résultats à la
semaine 30
| Sémaglutide 0,5 mg | Sémaglutide 1 mg | Placebo | |
| Population en intention de traiter (ITT) (n) | 128 | 130 | 129 |
| HbA1c (%) | |||
|
Inclusion
(moyenne)
|
8,1 | 8,1 | 8,0 |
|
Variation
entre l'inclusion et la semaine 30
|
-1,5 | -1,6 | 0 |
|
Différence
par rapport au placebo [IC 95 %]
|
-1,4 [-1,7 ; -1,1]a | -1,5 [-1,8 ; - 1,2]a | - |
| Patients (%) ayant atteint une HbA1c < 7 % | 74 | 72 | 25 |
| Glycémie à jeun (mmol/l) | |||
|
Inclusion
(moyenne)
|
9,7 | 9,9 | 9,7 |
|
Variation
entre l'inclusion et la semaine 30
|
-2,5 | -2,3 | -0,6 |
| Poids corporel (kg) | |||
|
Inclusion
(moyenne)
|
89,8 | 96,9 | 89,1 |
|
Variation
entre l'inclusion et la semaine 30
|
-3,7 | -4,5 | -1,0 |
|
Différence
par rapport au placebo [IC 95 %]
|
-2,7 [-3,9 ; -1,6]a | -3,6 [-4,7 ; - 2,4]a | - |
ap < 0,0001 (bilatéral) pour la supériorité
SUSTAIN
2 - Sémaglutide versus sitagliptine respectivement en association avec 1 ou 2
antidiabétiques oraux (metformine et/ou thiazolidinediones)
Lors d'un essai clinique contrôlé de 56 semaines en double aveugle versus
traitement actif, 1 231 patients ont été randomisés dans des groupes recevant
0,5 mg ou 1 mg de sémaglutide une fois par semaine ou 100 mg de sitagliptine
une fois par jour, tous en association avec de la metformine (94 %) et/ou des
thiazolidinediones (6 %).
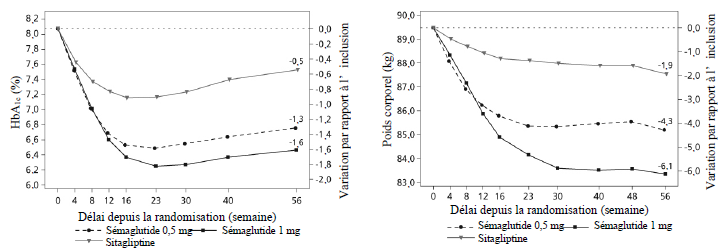
Tableau 3 SUSTAIN 2 : résultats à la
semaine 56
| Sémaglutide 0,5 mg | Sémaglutide 1 mg | Sitagliptine 100 mg | |
| Population en intention de traiter (ITT) (n) | 409 | 409 | 407 |
| HbA1c (%) | |||
|
Inclusion
(moyenne)
|
8,0 | 8,0 | 8,2 |
|
Variation
entre l'inclusion et la semaine 56
|
-1,3 | -1,6 | -0,5 |
|
Différence
par rapport à la sitagliptine [IC 95 %]
|
-0,8 [-0,9 ; -0,6]a | -1,1 [-1,2 ; - 0,9]a | - |
| Patients (%) ayant atteint une HbA1c < 7 % | 69 | 78 | 36 |
|
Glycémie
à jeun (mmol/l)
|
|||
|
Inclusion
(moyenne)
|
9,3 | 9,3 | 9,6 |
|
Variation
entre l'inclusion et la semaine 56
|
-2,1 | -2,6 | -1,1 |
| Poids corporel (kg) | |||
|
Inclusion
(moyenne)
|
89,9 | 89,2 | 89,3 |
|
Variation
entre l'inclusion et la semaine 56
|
-4,3 | -6,1 | -1,9 |
|
Différence
par rapport à la sitagliptine [IC 95 %]
|
-2,3 [-3,1 ; -1,6]a | -4,2 [-4,9 ; - 3,5]a | - |
ap < 0,0001 (bilatéral) pour la supériorité
SUSTAIN
7 - Sémaglutide versus dulaglutide respectivement en association avec la
metformine
Dans un essai ouvert de 40 semaines, 1 201 patients sous metformine ont été
randomisés 1 : 1 : 1 : 1 à 0,5 mg de sémaglutide, 0,75 mg de dulaglutide, 1 mg
de sémaglutide ou 1,5 mg de dulaglutide une fois par semaine respectivement.
L'essai a comparé 0,5 mg de sémaglutide à 0,75 mg de dulaglutide et 1mg de
sémaglutide à 1,5 mg de dulaglutide.
Les troubles
gastro-intestinaux ont été les effets indésirables les plus fréquents et se
sont produits de manière proportionnelle chez les patients sous sémaglutide 0,5
mg (129 patients [43 %]), sémaglutide 1 mg (133 [44 %]) et dulaglutide 1,5 mg
(143 [48 %]) ; moins de patients avaient de troubles gastro- intestinaux sous
dulaglutide 0,75 mg (100 [33 %]).
À la semaine 40, l'augmentation de la fréquence cardiaque sous sémaglutide (0,5
mg et 1 mg) et sous dulaglutide (0,75 mg et 1,5 mg) était de 2,4 ; 4,0 et 1,6 ;
2,1 battements/min, respectivement.
Tableau 4 SUSTAIN 7 : résultats à la
semaine 40
| Sémaglutide 0,5 mg | Sémaglutide 1 mg | Dulaglutide 0,75 mg | Dulaglutide 1,5 mg | |
| Population en intention de traiter (ITT) (n) | 301 | 300 | 299 | 299 |
| HbA1c (%) | ||||
|
Inclusion
(moyenne)
|
8,3 | 8,2 | 8,2 | 8,2 |
|
Variation
entre l'inclusion et la semaine 40
|
-1,5 | -1,8 | -1,1 | -1,4 |
|
Différence
par rapport au dulaglutide
[IC 95 %]
|
-0,4b [-0,6 ; -0,2]a |
-0,4c [-0,6 ; -0,3]a |
- | - |
| Patients (%) ayant atteint une HbA1c < 7 % | 68 | 79 | 52 | 67 |
| Glycémie à jeun (mmol/l) | ||||
|
Inclusion
(moyenne)
|
9,8 | 9,8 | 9,7 | 9,6 |
|
Variation
entre l'inclusion et la semaine 40
|
-2,2 | -2,8 | -1,9 | -2,2 |
| Poids corporel (kg) | ||||
|
Inclusion
(moyenne)
|
96,4 | 95,5 | 95,6 | 93,4 |
|
Variation
entre l'inclusion et la semaine 40
|
-4,6 | -6,5 | -2,3 | -3,0 |
|
Différence
par rapport au dulaglutide
[IC 95 %]
|
-2,3b [-3,0 ; -1,5]a |
-3,6c [-4,3 ; -2,8]a |
- | - |
ap <0,0001 (bilatéral) pour la supériorité
b sémaglutide 0,5 mg vs dulaglutide 0,75 mg
c sémaglutide 1 mg vs dulaglutide 1,5 mg
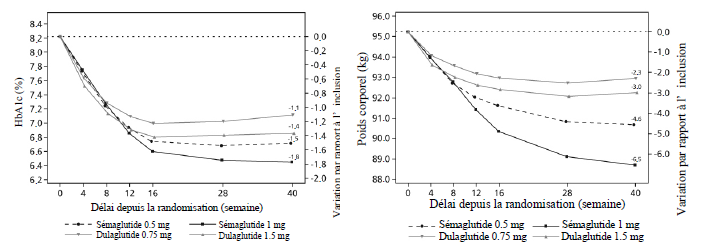
Figure 2 Variation moyenne de l'HbA1c (%) et du poids corporel (kg) entre l'inclusion et la semaine 40
b sémaglutide 0,5 mg vs dulaglutide 0,75 mg
c sémaglutide 1 mg vs dulaglutide 1,5 mg
Figure 2 Variation moyenne de l'HbA1c (%) et du poids corporel (kg) entre l'inclusion et la semaine 40
SUSTAIN 3 - Sémaglutide versus exénatide à libération
prolongée respectivement en association avec la metformine ou la metformine
plus sulfamide hypoglycémiant
Dans un essai en ouvert de 56 semaines, 813 patients sous metformine seule (49
%), metformine plus sulfamide hypoglycémiant (45 %) ou autre (6 %) ont été
randomisés dans des groupes recevant 1 mg de sémaglutide ou 2 mg d'exénatide à
libération prolongée une fois par semaine.
Tableau 5 SUSTAIN 3 : résultats à la
semaine 56
| Sémaglutide 1 mg | Exénatide à libération prolongée 2 mg | |
| Population en intention de traiter (ITT) (n) | 404 | 405 |
| HbA1c (%) | ||
|
Inclusion
(moyenne)
|
8,4 | 8,3 |
|
Variation
entre l'inclusion et la semaine 56
|
-1,5 | -0,9 |
|
Différence
par rapport à l'exénatide [IC 95 %]
|
-0,6 [-0,8 ; -0,4]a | - |
| Patients (%) ayant atteint une HbA1c < 7 % | 67 | 40 |
| Glycémie à jeun (mmol/l) | ||
|
Inclusion
(moyenne)
|
10,6 | 10,4 |
|
Variation
entre l'inclusion et la semaine 56
|
-2,8 | -2,0 |
| Poids corporel (kg) | ||
|
Inclusion
(moyenne)
|
96,2 | 95,4 |
|
Variation
entre l'inclusion et la semaine 56
|
-5,6 | -1,9 |
|
Différence
par rapport à l'exénatide [IC 95 %]
|
-3,8 [-4,6 ; -3,0]a | - |
ap < 0,0001 (bilatéral) pour la supériorité
SUSTAIN
4 - Sémaglutide versus insuline glargine respectivement en association avec 1
ou 2 antidiabétiques oraux (metformine ou metformine et sulfamide
hypoglycémiant)
Dans un essai comparateur en ouvert de 30 semaines, 1 089 patients ont été
randomisés dans des groupes recevant 0,5 mg ou 1 mg de sémaglutide une fois par
semaine ou de l'insuline glargine une fois par jour en association avec un
traitement de fond par metformine (48 %) ou metformine et sulfamide hypoglycémiant
(51 %).
Tableau 6 SUSTAIN 4 : résultats à la
semaine 30
| Sémaglutide 0,5 mg | Sémaglutide 1 mg | Insuline glargine | |
| Population en intention de traiter (ITT) (n) | 362 | 360 | 360 |
| HbA1c (%) | |||
|
Inclusion
(moyenne)
|
8,1 | 8,2 | 8,1 |
|
Variation
entre l'inclusion et la semaine 30
|
-1,2 | -1,6 | -0,8 |
|
Différence
par rapport à l'insuline glargine [IC 95 %]
|
-0,4 [-0,5 ; -0,2]a | -0,8 [-1,0 ; -0,7]a | - |
| Patients (%) ayant atteint une HbA1c < 7 % | 57 | 73 | 38 |
| Glycémie à jeun (mmol/l) | |||
|
Inclusion
(moyenne)
|
9,6 | 9,9 | 9,7 |
|
Variation
entre l'inclusion et la semaine 30
|
-2,0 | -2,7 | -2,1 |
| Poids corporel (kg) | |||
|
Inclusion
(moyenne)
|
93,7 | 94,0 | 92,6 |
|
Variation
entre l'inclusion et la semaine 30
|
-3,5 | -5,2 | +1,2 |
|
Différence
par rapport à l'insuline glargine [IC 95 %]
|
-4,6 [-5,3 ; -4,0]a | -6,34 [-7,0 ; -5,7]a | - |
ap < 0,0001 (bilatéral) pour la supériorité
SUSTAIN
5 - Sémaglutide versus placebo respectivement en association avec l'insuline
basale
Lors d'un essai clinique contrôlé de 30 semaines en double aveugle versus
placebo, 397 patients insuffisamment contrôlés par insuline basale avec ou sans
metformine ont été randomisés dans des groupes recevant 0,5 mg ou 1 mg de
sémaglutide une fois par semaine ou un placebo.
Tableau 7 SUSTAIN 5 : résultats à la
semaine 30
| Sémaglutide 0,5 mg | Sémaglutide 1 mg | Placebo | |
| Population en intention de traiter (ITT) (n) | 132 | 131 | 133 |
| HbA1c (%) | |||
|
Inclusion
(moyenne)
|
8,4 | 8,3 | 8,4 |
|
Variation
entre l'inclusion et la semaine 30
|
-1,4 | -1,8 | -0,1 |
|
Différence
par rapport au placebo [IC 95 %]
|
-1,4 [-1,6 ; -1,1]a | -1,8 [-2,0 ; - 1,5]a | - |
| Patients (%) ayant atteint une HbA1c < 7 % | 61 | 79 | 11 |
| Glycémie à jeun (mmol/l) | |||
|
Inclusion
(moyenne)
|
8,9 | 8,5 | 8,6 |
|
Variation
entre l'inclusion et la semaine 30
|
-1,6 | -2,4 | -0,5 |
| Poids corporel (kg) | |||
|
Inclusion
(moyenne)
|
92,7 | 92,5 | 89,9 |
|
Variation
entre l'inclusion et la semaine 30
|
-3,7 | -6,4 | -1,4 |
|
Différence
par rapport au placebo [IC 95 %]
|
-2,3 [-3,3 ; -1,3]a | -5,1 [-6,1 ; - 4,0]a | - |
ap < 0,0001 (bilatéral) pour la supériorité
SUSTAIN
FORTE - Sémaglutide 2 mg versus sémaglutide 1 mg
Lors d'un essai de 40 semaines en double aveugle, 961 patients insuffisamment
contrôlés par metformine avec ou sans sulfamide hypoglycémiant ont été
randomisés dans des groupes recevant 2 mg ou 1 mg de sémaglutide une fois par
semaine.
Le traitement par sémaglutide de 2 mg a résulté en une réduction statistiquement supérieure de l'HbA1c après 40 semaines de traitement par rapport au sémaglutide de 1 mg.
Tableau 8 SUSTAIN FORTE : résultats
à la semaine 40
| Sémaglutide 1 mg | Sémaglutide 2 mg | |
| Population en intention de traiter (ITT) (n) | 481 | 480 |
| HbA1c (%) | ||
|
Inclusion
(moyenne)
|
8,8 | 8,9 |
|
Variation
entre l'inclusion et la semaine 40
|
-1,9 | -2,2 |
|
Différence
par rapport au sémaglutide à 1 mg [IC 95 %]
|
- | -0,2 [-0,4, -0,1]a |
| Patients (%) ayant atteint une HbA1c < 7 % | 58 | 68 |
| Glycémie à jeun (mmol/l) | ||
|
Inclusion
(moyenne)
|
10,9 | 10,7 |
|
Variation
entre l'inclusion et la semaine 40
|
-3,1 | -3,4 |
| Poids corporel (kg) | ||
|
Inclusion
(moyenne)
|
98,6 | 100,1 |
|
Variation
entre l'inclusion et la semaine 40
|
-6,0 | -6,9 |
|
Différence
par rapport au sémaglutide à 1 mg [IC 95 %]
|
- | -0,9 [-1,7, -0,2]b |
ap < 0,001 (bilatéral) pour la supériorité
bp < 0,05 (bilatéral) pour la supériorité
bp < 0,05 (bilatéral) pour la supériorité
SUSTAIN
9 - Sémaglutide versus placebo en complément d'un inhibiteur du SGLT2 ±
metformine ou sulfamide hypoglycémiant
Lors d'un essai clinique contrôlé de 30 semaines en double aveugle versus placebo,
302 patients insuffisamment contrôlés par un inhibiteur du SGLT2 avec ou sans
metformine ou sulfamide hypoglycémiant ont été randomisés dans des groupes
recevant 1,0 mg de sémaglutide une fois par semaine ou un placebo.
Tableau 9 SUSTAIN 9 : résultats à la
semaine 30
| Sémaglutide 1 mg | Placebo | |
| Population en intention de traiter (ITT) (n) | 151 | 151 |
| HbA1c (%) | ||
|
Inclusion
(moyenne)
|
8,0 | 8,1 |
|
Variation
entre l'inclusion et la semaine 30
|
-1,5 | -0,1 |
|
Différence
par rapport au placebo [IC 95 %]
|
-1,4 [-1,6 ; -1,2]a | - |
| Patients (%) ayant atteint une HbA1c < 7 % | 78,7 | 18,7 |
| Glycémie à jeun (mmol/l) | ||
|
Inclusion
(moyenne)
|
9,1 | 8,9 |
|
Variation
entre l'inclusion et la semaine 30
|
-2,2 | 0,0 |
| Poids corporel (kg) | ||
|
Inclusion
(moyenne)
|
89,6 | 93,8 |
|
Variation
entre l'inclusion et la semaine 30
|
-4,7 | -0,9 |
|
Différence
par rapport au placebo [IC 95 %]
|
-3,8 [-4,7 ; -2,9]a | - |
ap < 0,0001 (bilatéral) pour la supériorité, après
ajustement pour multiplicité sur la base de tests hiérarchiques de la valeur de
l'HbA1c et du poids corporel
SUSTAIN
11 - Sémaglutide versus insuline asparte en association à l'insuline glargine +
metformine
Lors d'un essai en ouvert de 52 semaines, 1 748 sujets présentant un diabète de
type 2, insuffisamment contrôlé après une phase préliminaire de 12 semaines
sous insuline glargine et metformine, ont été randomisés en 1:1 pour recevoir
soit du sémaglutide une fois par semaine (0,5 mg ou 1,0 mg), soit de l'insuline
asparte trois fois par jour. La population incluse avait un diabète de 13,4 ans
en moyenne et une HbA1c de 8,6 % en moyenne, avec une HbA1c cible de 6,5 - 7,5
%.
Le traitement par sémaglutide a entraîné une réduction de l'HbA1c en semaine 52 (-1,5 % pour le sémaglutide versus -1,2 % pour l'insuline asparte).
Le nombre d'épisodes hypoglycémiques sévères dans les deux bras de traitement était faible (4 épisodes avec sémaglutide versus 7 épisodes avec insuline asparte).
Le poids corporel initial moyen a diminué avec le sémaglutide (-4,1 kg) et a augmenté avec l'insuline asparte (+2,8 kg), et la différence estimée entre traitements était de -6,99 kg (IC à 95 % : -7,41 à -6,57) en semaine 52.
Association
avec un sulfamide hypoglycémiant en monothérapie
Dans SUSTAIN 6 (voir sous-rubrique « Maladies cardiovasculaires »), 123
patients prenaient un sulfamide hypoglycémiant en monothérapie à l'inclusion.
L'HbA1c à l'inclusion était de 8,2 %, 8,4 % et 8,4 % pour les groupes 0,5 mg de
sémaglutide, 1 mg de sémaglutide et placebo, respectivement. À la semaine 30,
la variation de l'HbA1c était de -1,6 %, -1,5 % et 0,1 % dans les groupes 0,5
mg de sémaglutide, 1 mg de sémaglutide et placebo, respectivement.
Association
avec de l'insuline prémélangée ± 1-2 ADO
Dans SUSTAIN 6 (voir sous-rubrique Maladies cardiovasculaires), 867 patients
étaient sous insuline prémélangée (avec ou sans ADO) à l'inclusion. L'HbA1c à
l'inclusion était de 8,8 %, 8,9 % et 8,9 % pour les groupes 0,5 mg de
sémaglutide, 1 mg de sémaglutide et placebo, respectivement. À la semaine 30,
la variation de l'HbA1c était de -1,3 %, -1,8 % et -0,4 % dans les groupes 0,5
mg de sémaglutide, 1 mg de sémaglutide et placebo, respectivement.
Maladies
cardiovasculaires
Lors d'un essai en double aveugle de 104 semaines (SUSTAIN 6), 3 297 patients
diabétiques de type 2 à haut risque cardiovasculaire ont été randomisés dans
des groupes recevant 0,5 mg ou 1 mg de sémaglutide une fois par semaine ou un
placebo correspondant, tous en association au traitement standard avec un suivi
de 2 ans. Au total, 98 % des patients ont terminé l'essai, et le statut vital
de 99,6 % des patients était connu à la fin de l'essai.
La répartition démographique de la population de l'essai était la suivante : 1 598 patients (48,5 %) ≥ 65 ans, 321 (9,7 %) ≥ 75 ans et 20 (0,6 %) ≥ 85 ans. 2 358 patients présentaient une fonction rénale normale ou une légère insuffisance rénale, 832 une insuffisance rénale modérée et 107, une insuffisance rénale sévère ou au stade terminal. Les patients comptaient 61 % d'hommes, l'âge moyen était de 65 ans et l'IMC moyen de 33 kg/m2. L'ancienneté moyenne du diabète était de 13,9 ans.
Le critère primaire était le délai de survenue depuis la randomisation du premier événement cardiovasculaire majeur (MACE) : mortalité cardiovasculaire, infarctus du myocarde non-fatal ou accident vasculaire cérébral non-fatal.
Le nombre total de composants primaires des critères MACE était de 254, dont 108 (6,6 %) avec le sémaglutide et 146 (8,9 %) avec un placebo. Voir figure 4 pour des résultats concernant les critères cardiovasculaires primaires et secondaires. Le traitement par sémaglutide a entraîné une réduction de 26 % du risque concernant le critère composite primaire : mortalité cardiovasculaire, infarctus du myocarde non-fatal ou accident vasculaire cérébral non-fatal. Le nombre total de décès cardiovasculaires, d'infarctus du myocarde non-fatals et d'accidents vasculaires cérébraux non-fatals était de 90, 111, et 71, respectivement, dont 44 (2,7 %), 47 (2,9 %), et 27 (1,6 %), respectivement, sous sémaglutide (figure 4). La réduction du risque concernant le critère composite primaire était principalement due à une baisse du taux d'accidents vasculaires cérébraux non-fatals (39 %) et d'infarctus du myocarde non-fatals (26 %) (figure 3).

Figure 3 Représentation Kaplan-Meier du délai de survenue du premier
événement composite : mortalité cardiovasculaire, infarctus du myocarde
non-fatal ou accident vasculaire cérébral non-fatal (SUSTAIN 6)

Figure 4 Graphique en forêt : analyses du délai de survenue du premier événement composite, de ses composants et de mortalité toutes causes confondues (SUSTAIN 6)
Il a été observé 158 événements d'apparition ou d'aggravation de néphropathie. Le hazard ratio [IC 95 %] concernant le délai d'apparition de la néphropathie (apparition d'une macroalbuminurie persistante, doublement persistant de la créatinine sérique, besoin d'une thérapie de remplacement rénal continue et décès dû à une maladie rénale) était de 0,64 [0,46 ; 0,88], s'expliquant par l'apparition d'une macroalbuminurie persistante.
Résultats rénaux
Lors d’un essai en double aveugle évaluant les effets rénaux (FLOW), 3
533 patients atteints de diabète de type 2 et de maladie rénale
chronique avec un DFGe compris entre 50 et 75 ml/min/1,73 m2 et un RAC > 300 et < 5 000 mg/g ou un DFGe compris entre 25 et < 50 ml/min/1,73 m2
et un RAC > 100 et < 5 000 mg/g ont été randomisés dans des
groupes recevant 1 mg de sémaglutide une fois par semaine ou un placebo
correspondant, en association au traitement standard.
L’étude a été arrêtée précocement pour efficacité après l’analyse
intermédiaire planifiée sur la base d’une recommandation du comité
indépendant de surveillance des données. Le suivi médian était de 40,9
mois.
L’âge moyen de la population était de 66,6 ans et les patients
comptaient 69,7 % d’hommes. L’IMC initial moyen était de 32,0 kg/m2. L’ancienneté moyenne du diabète à l’inclusion était de 17,4 ans et l’HbA1c initial moyen était de 7,8 % (61,5 mmol/mol). Le DFGe initial moyen était de 47 ml/min/1,73 m2
et le RAC médian était de 568 mg/g. À l’inclusion, environ 95 % des
patients étaient traités par inhibiteurs du système
rénine-angiotensine-aldostérone et 16 % par inhibiteurs du
co-transporteur de sodium-glucose de type 2 (SGLT2).
Le sémaglutide a été supérieur au
placebo, en association au traitement standard, en termes de prévention
du critère composite primaire : réduction persistante ≥ 50 % du DFGe,
apparition d’un DFGe persistant < 15 ml/min/1,73 m2,
initiation d’un traitement de suppléance rénale chronique, mortalité
rénale ou mortalité cardiovasculaire avec un hazard ratio de 0,76 [IC
95 % : 0,66 ; 0,88] correspondant à une réduction du risque relatif de
progression de la maladie rénale de 24 % (voir figure 5). Les
composants individuels du critère composite primaire ont contribué à
l’effet du traitement mais les décès d’origine rénale ont été peu
nombreux (voir figure 6).
Le sémaglutide s’est révélé supérieur au placebo, en association au
traitement standard, en termes de réduction du taux annuel de variation
du DFGe, avec une différence estimée entre les traitements de 1,16
ml/min/1,73 m2/an [IC 95 % ; 0,86 ; 1,47]. Le traitement par
sémaglutide a amélioré la survie globale avec une réduction
significative de la mortalité toutes causes confondues (voir figure 6).
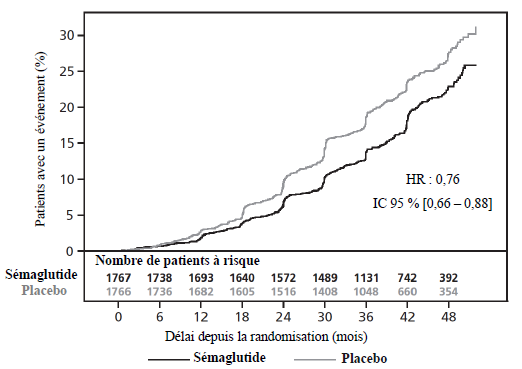
Figure 5 Fonction d’incidence cumulée du délai de survenue du premier événement du critère composite primaire : apparition d’une réduction persistante ≥ 50 % du DFGe, apparition d’un DFGe persistant < 15 ml/min/1,73 m2, initiation d’un traitement de suppléance rénale chronique, mortalité rénale ou cardiovasculaire (FLOW)
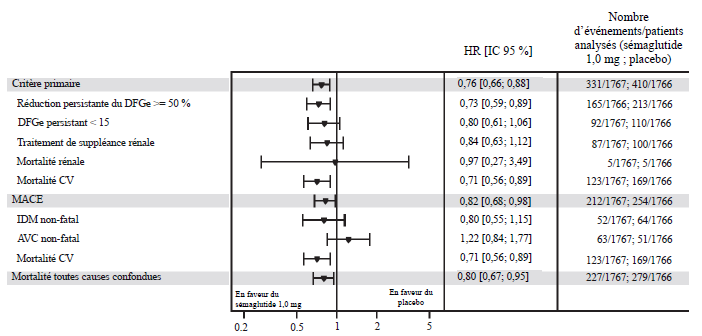
Figure
6 Graphique en forêt : analyses du délai de survenue du premier
événement du critère composite primaire et de ses composants, du
premier MACE et de ses composants et de mortalité toutes causes
confondues (FLOW)
Poids
corporel
Après un an de traitement, une perte de poids de ≥ 5 % et ≥ 10 % a
été obtenue chez davantage de patients sous 0,5 mg (46 % et 13 %) et 1 mg de
sémaglutide (52 - 62 % et 21 - 24 %) par rapport aux comparateurs actifs
sitagliptine (18 % et 3 %) et exénatide à libération prolongée (17 % et 4 %).
Dans un essai de 40 semaines versus dulaglutide, une perte de poids ≥ 5 % et ≥ 10 % a été obtenue chez davantage de patients sous sémaglutide 0,5 mg (44 % et 14 %) par rapport au dulaglutide 0,75 mg (23 % et 3 %) et sous sémaglutide 1 mg (jusqu'à 63 % et 27 %) par rapport au dulaglutide 1,5 mg (30 % et 8 %).
Une réduction significative et durable du poids corporel entre l'inclusion et la semaine 104 a été observée avec 0,5 mg et 1 mg de sémaglutide vs 0,5 mg et 1 mg de placebo, en association avec un traitement standard (-3,6 kg et -4,9 kg vs -0,7 kg et -0,5 kg, respectivement) dans l'étude SUSTAIN 6.
Lors de l’essai FLOW évaluant les
effets rénaux, le traitement par 1 mg de sémaglutide a entraîné une
réduction durable du poids corporel à la semaine 104 vs placebo, en
association avec un traitement standard (-5,6 kg avec le sémaglutide et
-1,4 kg avec le placebo).
Pression
artérielle
Des réductions significatives de la pression artérielle systolique moyenne ont
été observées en cas d'utilisation de 0,5 mg (3,5 - 5,1 mmHg) et de 1 mg de
sémaglutide (5,4 - 7,3 mmHg) en association avec des antidiabétiques oraux ou
de l'insuline basale. Pour la pression artérielle diastolique, aucune
différence significative n'a été constatée entre le sémaglutide et les
comparateurs. Les réductions observées concernant la pression artérielle
systolique pour le sémaglutide de 2 mg et de 1 mg à la semaine 40 étaient de
-5,3 mmHg et de -4,5 mmHg, respectivement.
Population pédiatrique
L'Agence européenne des médicaments a différé l'obligation de soumettre les résultats d'études réalisées avec Ozempic dans un ou plusieurs sous-groupes de la population pédiatrique diabétique de type 2 (voir rubrique Posologie et mode d'administration pour des informations sur l'utilisation dans la population pédiatrique).
En comparaison avec le GLP-1 natif, le sémaglutide a une demi-vie prolongée d'environ une semaine, ce qui permet de l'administer en sous-cutanée une fois par semaine. Le principal mécanisme d'action prolongée est la liaison à l'albumine, qui entraîne une baisse de la clairance rénale et une protection contre la dégradation métabolique. De plus, le sémaglutide est stabilisé de manière à éviter la dégradation par l'enzyme DPP-4.
Absorption
La concentration maximale a été atteinte 1 à 3 jours après administration de la dose. L'exposition à l'état d'équilibre a été atteinte après 4 à 5 semaines d'administration une fois par semaine. Chez les patients diabétiques de type 2, les concentrations moyennes à l'état d'équilibre après administration sous-cutanée de 0,5 mg et 1 mg de sémaglutide étaient d'environ 16 nmol/l et 30 nmol/l, respectivement. Dans l'étude comparant le sémaglutide de 1 mg et de 2 mg, les concentrations moyennes à l'état d'équilibre étaient de 27 nmol/l et de 54 nmol/l, respectivement. L'exposition au sémaglutide a augmenté de manière dose-proportionnelle pour des doses de 0,5 mg, 1 mg et 2 mg. Une exposition similaire a été obtenue avec une administration sous-cutanée de sémaglutide dans l'abdomen, la cuisse ou le haut du bras. La biodisponibilité absolue du sémaglutide par voie sous- cutanée était de 89 %.
Distribution
Le volume de distribution moyen du sémaglutide après administration sous-cutanée chez des patients diabétiques de type 2 était d'environ 12,5 l. Le sémaglutide était fortement lié à l'albumine plasmatique (> 99 %).
Biotransformation
Avant l'excrétion, le sémaglutide est fortement métabolisé par clivage protéolytique de la chaîne peptidique et bêta-oxydation séquentielle de la chaîne latérale des acides gras. L'enzyme endopeptidase neutre (EPN) interviendrait dans le métabolisme du sémaglutide.
Élimination
Dans
un essai utilisant une dose sous-cutanée unique de sémaglutide
radiomarqué, il a été observé que les voies d'excrétion principales des
métabolites du sémaglutide étaient via l'urine et les fèces ; environ
2/3 des métabolites du sémaglutide étaient excrétés dans l'urine et
environ 1/3 dans les fèces. Approximativement 3 % de la dose a été
excrétée sous la forme de sémaglutide intact dans l'urine.
Chez des patients diabétiques de type 2, la clairance du sémaglutide
était d'environ 0,05 l/h. Avec une demi-vie d'élimination d'environ 1
semaine, le sémaglutide restera présent dans la circulation pendant
approximativement 5 semaines après la dernière dose.
Population particulière
Sujets âgés
L'âge n'a aucun effet sur la pharmacocinétique du sémaglutide, selon
les données des études de phase 3a portant sur des patients de 20 à 86
ans.
Genre et origine ethnique
Le genre et l'origine ethnique (Blanc, Noir, Afro-Américain, Asiatique,
Hispanique ou Latino, non- Hispanique ou non-Latino) n'ont eu aucun
effet sur la pharmacocinétique du sémaglutide.
Poids corporel
Le poids corporel influence l'exposition au sémaglutide. Un poids
corporel plus élevé diminue l'exposition ; une différence de 20 % de
poids corporel entre les patients entraîne une différence d'environ 16
% de l'exposition. Des doses de sémaglutide de 0,5 mg et de 1 mg
assurent une exposition systémique adéquate à un poids corporel compris
entre 40 et 198 kg.
Insuffisance rénale
L'insuffisance rénale n'a pas affecté la pharmacocinétique du
sémaglutide de manière cliniquement significative. Cela a été observé
avec une dose unique de 0,5 mg de sémaglutide chez des patients
présentant des degrés divers d'insuffisance rénale (légère, modérée,
sévère ou patients dialysés) en comparaison avec des sujets à la
fonction rénale normale. Cela a également été observé chez des patients
diabétiques de type 2 présentant une insuffisance rénale sur la base de
données d'études de phase 3a, bien que l'expérience ait été limitée
chez des patients atteints de maladie rénale au stade terminal.
Insuffisance hépatique
L'insuffisance hépatique n'a eu aucun impact sur l'exposition au
sémaglutide. La pharmacocinétique du sémaglutide a été évaluée chez des
patients présentant des degrés divers d'insuffisance hépatique (légère,
modérée, sévère) en comparaison avec des sujets présentant une fonction
hépatique normale dans le cadre d'un essai utilisant une dose unique de
0,5 mg de sémaglutide.
Population pédiatrique
Le sémaglutide n'a pas été étudié dans la population pédiatrique.
Immunogénicité
Des anticorps dirigés contre le sémaglutide lors d'un traitement par
sémaglutide de 1 mg ou de 2,4 mg sont apparus peu fréquemment (voir
rubrique Effets indésirables) et la réponse n'a pas semblée influencer la pharmacoccinétique du sémaglutide.
Le sémaglutide n'a aucun effet ou un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines. Lorsqu'il est utilisé en association à un sulfamide hypoglycémiant ou à une insuline, les patients doivent être informés qu'ils doivent prendre des précautions pour éviter une hypoglycémie lors de la conduite de véhicules ou de l'utilisation de machines (voir rubrique Mises en garde spéciales et précautions d'emploi).
Les données précliniques issues des études conventionnelles de pharmacologie de sécurité, toxicologie en administration répétée ou génotoxicité n'ont pas révélé de risque particulier pour l'homme.
Les tumeurs non létales des cellules C de la thyroïde observées chez les rongeurs constituent un effet spécifique à la classe des agonistes des récepteurs du GLP-1. Lors d'études de carcinogénicité sur 2 ans chez le rat et la souris, le sémaglutide a provoqué des tumeurs des cellules C de la thyroïde à des expositions cliniquement significatives. Aucun autre type de tumeurs liées au traitement n'a été observé. Les tumeurs des cellules C chez les rongeurs sont dues à un mécanisme non génotoxique, spécifique, médié par les récepteurs du GLP-1, auquel les rongeurs sont particulièrement sensibles. La pertinence de ces résultats pour l'homme est considérée comme faible mais ne peut pas être complètement exclue.
Lors d'études de fertilité chez le rat, le sémaglutide n'a pas affecté les performances d'accouplement ni la fertilité des mâles. Chez le rat femelle, une prolongation du cycle œstrien et une faible baisse du nombre d'ovulations ont été observées à des doses associées à une réduction du poids maternel.
Lors d'études du développement embryo-fœtal chez le rat, le sémaglutide a entraîné une embryotoxicité à des expositions inférieures aux niveaux cliniquement significatifs. Le sémaglutide a entraîné une nette réduction du poids maternel et une diminution de la croissance et de la survie embryonnaires. Chez les fœtus, des malformations viscérales et squelettiques majeures ont été observées, notamment des effets sur les os longs, les côtes, les vertèbres, la queue, les vaisseaux sanguins et les ventricules cérébraux. Des évaluations mécanistes ont indiqué que l'embryotoxicité impliquait une anomalie médiée par les récepteurs du GLP-1 au niveau de l'apport de nutriments à l'embryon via le sac vitellin du rat. En raison des différences d'anatomie et de fonction du sac vitellin entre les espèces, et en raison de l'absence d'expression des récepteurs du GLP-1 dans le sac vitellin des primates non humains, ce mécanisme n'est probablement pas pertinent chez l'homme. Cependant, un effet direct du sémaglutide sur le fœtus ne peut être exclu.
Lors d'études de toxicité pour le développement chez le lapin et le singe cynomolgus, une augmentation des fausses couches et une légère hausse de l'incidence des anomalies fœtales ont été observées à des expositions cliniquement significatives. Ces résultats coïncidaient avec une nette réduction du poids maternel allant jusqu'à 16 %. Il n'est pas établi si ces effets sont liés à la réduction de consommation d'aliments par la mère en tant qu'effet direct du GLP-1.
La croissance et le développement postnataux ont été évalués chez le singe cynomolgus. Les nourrissons étaient légèrement plus petits à la mise bas, mais ont récupéré pendant l'allaitement.
Chez les jeunes rats mâles et femelles, le sémaglutide a retardé la maturation sexuelle. Ces retards n'ont eu aucun impact sur la fertilité ou la capacité de reproduction des deux sexes, ni sur la capacité des femelles à maintenir une grossesse.
Le patient doit être averti du fait qu'il doit jeter l'aiguille après chaque injection et conserver le stylo sans aiguille attachée. Ceci pourra prévenir le risque d'obstruction des aiguilles, de contamination, d'infection, de fuite de la solution et de dose incorrecte.
Le stylo est réservé à l'utilisation par un seul patient.
Ozempic ne doit pas être utilisé si la solution n'est pas limpide et incolore ou presque incolore.
Ozempic ne doit pas être utilisé s'il a été congelé.
Ozempic peut être administré avec des aiguilles jetables de 30G, 31G et 32G d'une longueur maximale de 8 mm.
Tout médicament non utilisé et autres déchets doivent être éliminés conformément à la réglementation en vigueur.
Liste I.
Remboursement en fonction de l'indication (JO du 17/04/2019) :
Les seules indications thérapeutiques ouvrant droit à la prise en charge ou au remboursement par l'assurance maladie sont :
Chez les adultes pour le traitement du diabète de type 2 insuffisamment contrôlé en complément d'un régime alimentaire et d'une activité physique :
- en bithérapie en association à la metformine ;
- en trithérapie en association à la metformine et un sulfamide.
Extension d'indication remboursée (JO du 17/03/2023) :
- en trithérapie en association à la metformine et à l'insuline basale.
JO du 15/01/2025 :
La prise en charge de cette spécialité est subordonnée au renseignement par le prescripteur d'éléments relatifs aux circonstances et indications de la prescription en vue de l'établissement du document prévu au III de l'article R. 161-45 du code de la sécurité sociale.
Les éléments devant être renseignés par les prescripteurs sont les réponses aux questions suivantes :
- le patient est-il âgé de 18 ans ou plus ?
- le patient est-il atteint d'un diabète de type 2 insuffisamment contrôlé par un régime alimentaire et l'activité physique ?
- Ozempic® est-il prescrit dans l'une des situations suivantes :
- en association avec d'autres médicaments destinés au traitement du diabète ;
- en monothérapie, quand l'utilisation de la metformine est considérée comme inappropriée en raison d'une intolérance ou de contre-indications ?
Remboursement en fonction de l'indication (JO du 17/04/2019) :
Les seules indications thérapeutiques ouvrant droit à la prise en charge ou au remboursement par l'assurance maladie sont :
Chez les adultes pour le traitement du diabète de type 2 insuffisamment contrôlé en complément d'un régime alimentaire et d'une activité physique :
- en bithérapie en association à la metformine ;
- en trithérapie en association à la metformine et un sulfamide.
Extension d'indication remboursée (JO du 17/03/2023) :
- en trithérapie en association à la metformine et à l'insuline basale.
JO du 15/01/2025 :
La prise en charge de cette spécialité est subordonnée au renseignement par le prescripteur d'éléments relatifs aux circonstances et indications de la prescription en vue de l'établissement du document prévu au III de l'article R. 161-45 du code de la sécurité sociale.
Les éléments devant être renseignés par les prescripteurs sont les réponses aux questions suivantes :
- le patient est-il âgé de 18 ans ou plus ?
- le patient est-il atteint d'un diabète de type 2 insuffisamment contrôlé par un régime alimentaire et l'activité physique ?
- Ozempic® est-il prescrit dans l'une des situations suivantes :
- en association avec d'autres médicaments destinés au traitement du diabète ;
- en monothérapie, quand l'utilisation de la metformine est considérée comme inappropriée en raison d'une intolérance ou de contre-indications ?
Solution injectable (injection).
Solution isotonique, incolore ou presque incolore et limpide ; pH = 7,4.
Cartouche
de 3 ml (verre de type 1) fermée d'un côté par un piston en caoutchouc
(chlorobutyle) et de l'autre par un capuchon en aluminium avec un
opercule en caoutchouc stratifié (bromobutyle/polyisoprène) inséré. La
cartouche est assemblée dans un stylo prérempli jetable en
polypropylène, polyoxyméthylène, polycarbonate et acrylonitrile
butadiène styrène.
Présentation :
Ozempic 1 mg, solution injectable :
Chaque stylo prérempli contient 3 ml de solution, délivrant 4 doses de 1 mg.
1 stylo prérempli et 4 aiguilles NovoFine Plus à usage unique.
Un ml de solution contient 1,34 mg de sémaglutide*. Un stylo prérempli contient 4 mg de sémaglutide* dans 3 ml de solution. Chaque dose contient 1 mg de sémaglutide dans 0,74 ml de solution.
*Peptide analogue au glucagon-1 humain (GLP-1) produit par la technique de l'ADN recombinant sur des cellules de Saccharomyces cerevisiae.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Phosphate disodique dihydraté
Propylène glycol
Phénol
Acide chlorhydrique (pour l'ajustement du pH)
Hydroxyde de sodium (pour l'ajustement du pH)
Eau pour préparations injectables
Novo Nordisk
12 Cours Michelet
Carré Michelet
92800
Puteaux
Téléphone : 01 41 97 66 00
Information médicale
Tél
:
Ou 01 41
97 65 00 (Service et appel gratuits)
Fax : 
Email :
infomed@novonordisk.com
Site web : http://www.novonordisk.fr
Ozempic 2 mg, solution injectable n'est pas commercialisé en France.